


量子化学(quantum chemistry)是理论化学的一个分支学科,是应用量子力学的基本原理和方法研究化学问题的一门基础科学。从头算量子化学方法是基于量子化学的计算化学方法,是在给定原子核位置和电子数的情况下求解电子薛定谔方程,并以获得如电子密度、能量和热力学量等有用的信息的重要方法。从头计算电子结构方法旨在计算多电子的函数,即非相对论条件和玻恩-奥本海默近似下电子薛定谔方程的解。多电子函数通常是由多个简单电子函数的线性组合获得,主要函数是Hartree-Fock函数。在单电子近似的条件下,仅使用一个电子函数来近似上述多个简单函数,然后将单电子函数展开为有限基函数集的线性组合。研究范围包括稳定和不稳定分子的结构、性能及其结构与性能之间的关系;分子与分子之间的相互作用;分子与分子之间的相互碰撞和相互反应等问题。
研究方向:有机、无机、合成、小分子环境转化、团簇化学、均相催化、高分子等
计算体系:小分子、团簇、低聚物、自由基、离子等
常用软件:Gaussian,Gromacs,ORCA,Dmol3等
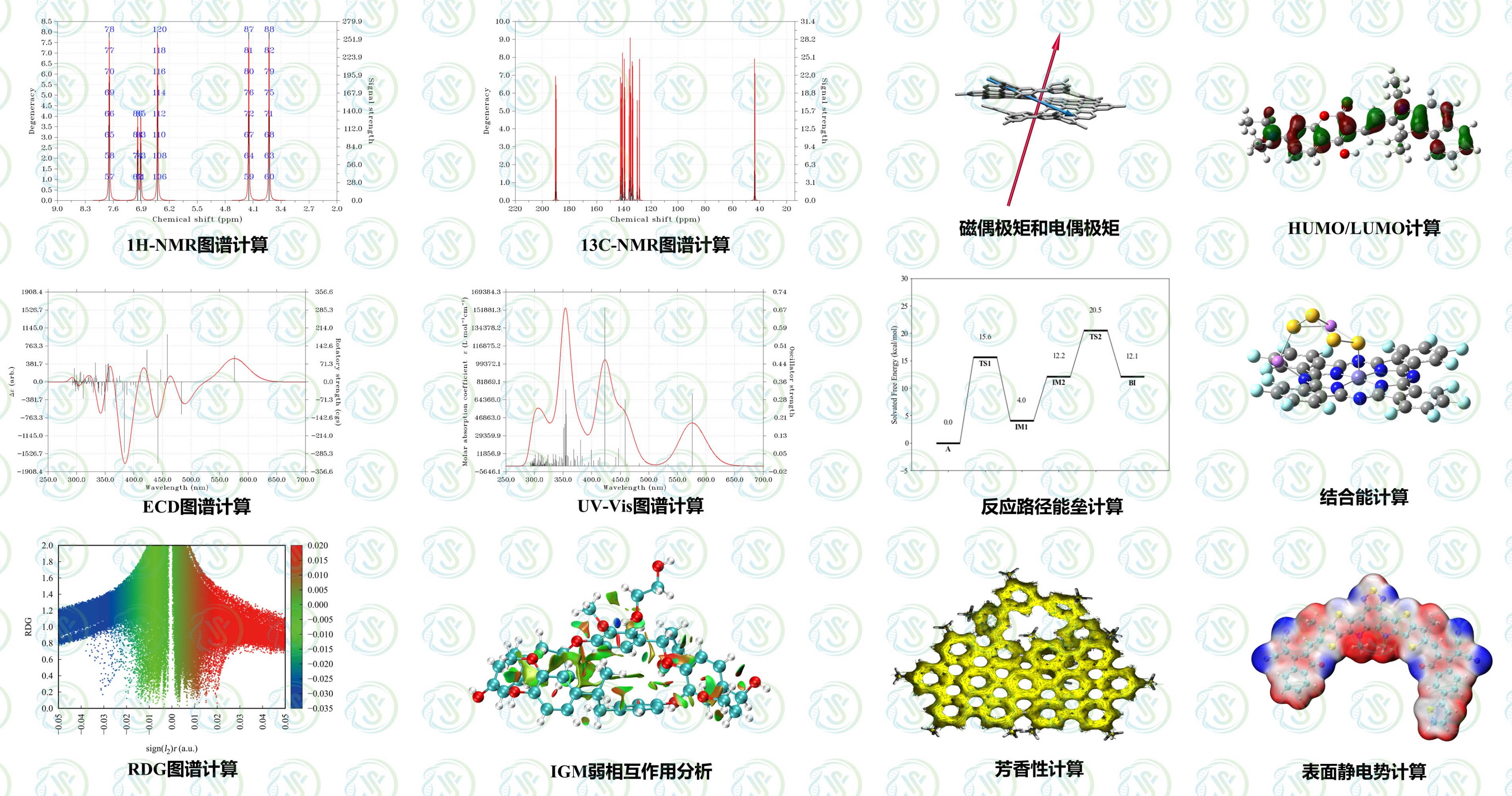
1、分子性质预测:静电势、偶极矩、布居数、轨道特性、键级、电荷、极化率、电子亲和能、电离势、自旋密度、电子转移等
2、化学反应机理:稳态及过渡态结构确定、反应热、反应能垒、反应机理及反应动力学等
3、激发态反应:激发态结构确定、激发能、跃迁偶极矩、荧光光谱、磷光光谱、势能面交叉研究等
4、弱相互作用:氢键、卤键、硫键、π-π堆积、盐桥、阳离子-π、疏水作用力等
5、光谱预测:红外、拉曼、紫外吸收、荧光、磷光、核磁谱、圆二色谱、旋光度等
1、先进的一站式数据分析解决方案
2、全球顶尖的专家团队
3、精尖的科研技术和手段
4、丰富的项目经验和发文经验
18817128943

扫码关注 森煜智研微信号

扫码关注 森煜智研公众号