


第一作者:Noemí Hernández-Haro
通讯作者:Antonio Frontera
通讯单位:Universitat de les Illes Balears;State Scientific-Production Enterprise “Istok”;Bogoliubov Laboratory of Theoretical Physics;Dubna State University
全文速览
该研究中Noemí等人利用密度泛函理论计算方法,对两种卤化钙钛矿进行了全面、系统的研究。研究了两种不同结构的钙钛矿,即立方CH3NH3PbI3 (MAPI)和HC(NH2)2PbI3 (FAPI)。使用了24个交换关联泛函,包括3个LDA泛函、10个GGA泛函、7个MGGA泛函和4个杂化泛函等进行了测试,以确定这些方法预测带隙的准确性。此外,还研究了处理钙钛矿计算的几种可能性。也就是说,Noemí等人已经用全电子相对论计算测试了数值原子轨道,并与具有赝势和相对论修正平面波框架进行了比较。此外,本文还研究了不同的k点集、赝势类型、有无单元优化、有无色散修正、有无自旋轨道耦合。结果表明,PBE和RPBE在计算精度和计算要求之间有较好的折衷。
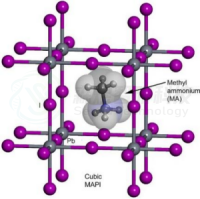
背景介绍
近年来,有机-无机金属卤化物钙钛矿太阳能电池(PSCs)成为光伏,纳米线和光磁研究的热点。这类由丰富的元素组成的材料其优点是化学和晶体学结构具有多功能性(如可调带隙)。此外,高吸收系数,长载流子扩散长度和低温处理也是此类材料的重要优点。值得注意的是,PSC的功率转换效率(PCE)超过22%。从结构上看,卤化物钙钛矿是由共角金属卤化物八面体构成,阳离子处在八面体之间的空隙(如图1)。三元卤化物钙钛矿的化学式为ABX3,其中A为阳离子(如CH3NH3+等),B为二价金属阳离子(如Pb2+,Sn2+),X为卤化物阴离子(如Cl−,Br− ,I−)。金属卤化物单元之间的相互作用会形成具有潜在的大带宽电子带。由于其固有的特性(低激子结合能和远距离载流子传输性质),卤化物钙钛矿已成为下一代太阳能电池最有希望的材料之一。人们普遍认为,其高的电子/空穴迁移率、高效的电子/空穴萃取和输运性能是PSCs高效的关键,而商业化仍需考虑其低成本、易加工和高稳定性。PSCs的长期稳定性是阻碍其商业化的另一个主要瓶颈。钙钛矿材料及其器件的退化机理目前尚不清楚。最近发表的一篇综述,总结了近年来在提高PSCs效率和稳定性方面所做的工作,并对今后的发展方向进行了有趣的讨论。
设计思路
为了从根本上了解卤化物钙钛矿的电子结构,包括其丰富的化学和物理性质以及这些性质之间的相互作用,人们对其进行了深入的理论研究。在这种情况下,一种能够可靠地计算材料特性(电子和热力学)的计算方法是必不可少的。标准DFT方法提供了钙钛矿可靠的结构和稳定性,但在许多情况下低估了带隙。另一方面,相对论GW近似已被证明可以精确地提供电子结构,但计算成本极高。因此,使用DFT计算仍是许多研究者预测设计钙钛矿性能的主要选择。应当指出,最近已证明DFT-1/2方法是准确的,计算费用很低。
数据介绍
在这项工作中,Noemí等人报告了一项全面和系统的DFT研究,使用了24个交换关联泛函,包括3个LDA泛函、10个GGA泛函、7个MGGA泛函和4个杂化泛函。并对这些泛函进行了测试,以确定这些方法预测MAPI和FAPI钙钛矿带隙的准确性。也同样系统的比较了数值原子轨道(NAO)与具有赝势和相对论校正的平面波基组。此外还测试了不同的k点集、赝势类型、单元优化、色散校正和自旋轨道耦合(SOC)。
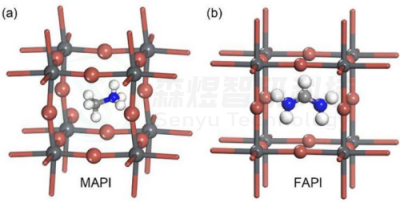
图2 MAPI和FAPI立方结构图
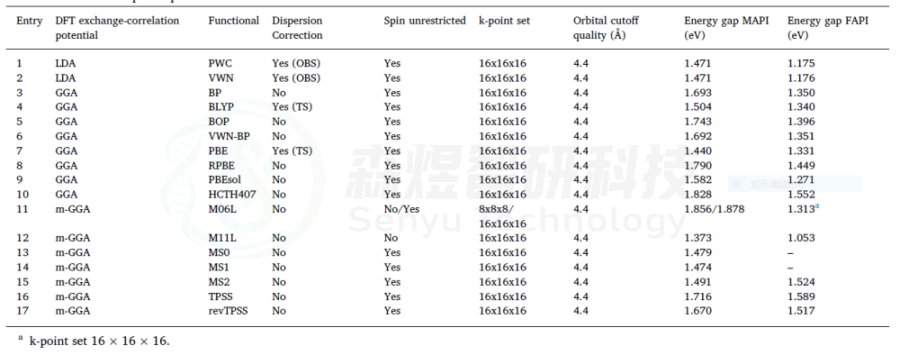
表1使用DMol3软件计算MAPI和FAPI的带隙。所有情况都使用了DNP基集,收敛标准设置为2.0·10-5Ha。在所有的情况下都使用了电子相对论赝势。
表2使用CASTEP软件计算MAPI和FAPI的带隙。收敛标准设置为2.0·10−6 Ha。所有情况都使用了Koelling-Hamon相对论。1-6使用了OTFG超软赝势类型。7-12使用了模守恒赝势。
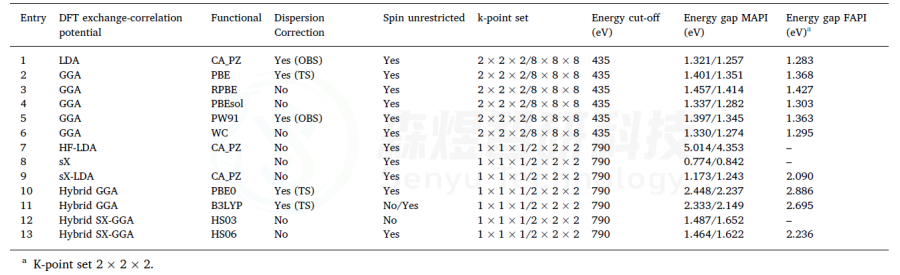
表3 利用CASTEP软件计算了MAPI和FAPI的带隙值,并考虑了自旋轨道耦合(SOC)效应。收敛标准设为2.0·10−6。所有情况都使用了Koelling-Hamon相对论,并采用模守恒赝势。所有计算都未约束。
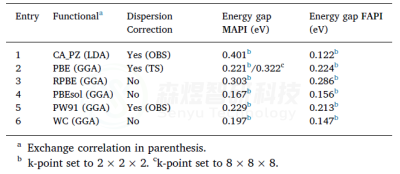
表4MAPI的结构优化参数a,b,c和α,β,γ(°),使用CASTEP和不同泛函计算,没有考虑SOC。收敛标准设置为2.0·10−6 Ha,使用Koelling-Hamon方法进行相对论处理。在所有情况下,计算是不受限制,自旋和k点设置为2×2×2。
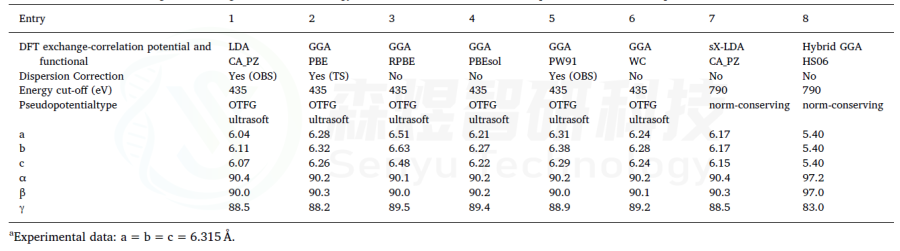
表5 FAPI的结构优化参数a,b,c和α,β,γ(°),使用CASTEP和不同泛函计算,没有考虑SOC。收敛标准设置为2.0·10−6 Ha,使用Koelling-Hamon方法进行相对论处理。在所有情况下,计算是不受限制,自旋和k点设置为2×2×2。
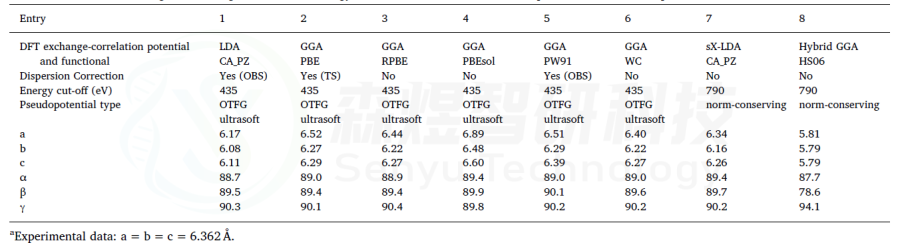
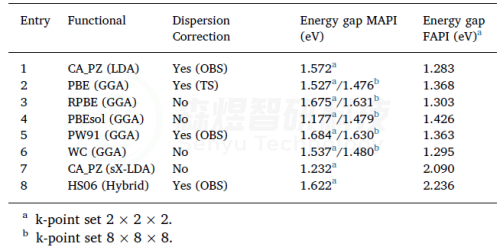
图6使用CASTEP计算MAPI和FAPI的带隙值。收敛标准设置为2.0·10−6 Ha。使用Koelling-Hamon方法进行相对论处理。1-6截断能设置为435,7-8设置为790。所有的计算都未限制自旋
总结
本文进行了一个全面的理论研究,其中使用了两个不同的代码。DMol3包括NAOs (DNP)和全电子相对论计算,另一个(CASTEP)使用平面波赝势法和相对论修正。在后者中,可以考虑SOC。从这项工作的结果可以得出以下结论:
1.利用NAOs代替平面波提高了理论与实验的一致性。
2.一般来说,GGA纯函数提供了对带隙的合理估计。
3.SOC的加入导致了对带隙严重低估(∼1 eV)。
4.PBE和RPBE是计算精度和计算需求之间的良好折衷。
5.不推荐使用混合泛函,特别是Minnesota泛函,因为考虑到与纯泛函相比计算开销较大。
论文信息
Noemí Hernández-Haro , Joaquín Ortega-Castro , Yaroslav B. Martynov , Rashid G. Nazmitdinov , Antonio Frontera,Universitat de les Illes Balears, Department of Chemistry, Crta. de Valldemossa km 7.5, 07122 Palma de Mallorca (Baleares), Spain,State Scientific-Production Enterprise “Istok”, 141120 Fryazino, Russia,Bogoliubov Laboratory of Theoretical Physics, Joint Institute for Nuclear Research, 141980 Dubna, Russia,Dubna State University, 141982 Dubna, Moscow Region, Russia,DFT prediction of band gap in organic-inorganic metal halide perovskites: An exchange-correlation functional benchmark study,Received 17 August 2018, Accepted 17 September 2018, Available online 18 September 2018, Version of Record 25 September 2018.

